

News|Articles|April 13, 2026
Case Report Identifies Type 1 Hereditary Angioedema in the Context of Omalizumab-Responsive CSU
Author(s)Marie Bosslett, Assistant Editor
Listen
0:00 / 0:00
Key Takeaways
- Omalizumab effectively controlled wheals and pruritus with improved UCT and DLQI, while recurrent, short-lived, nonpruritic angioedema persisted, indicating a distinct, non–mast cell–mediated process.
- Markedly reduced C4 and C1-INH levels, plus SERPING1 c.991C>A (p.P331T), established type 1 HAE despite absent family history, consistent with possible de novo mutation.
A rare case shows HAE with chronic spontaneous urticaria; non-itchy facial swelling despite omalizumab signals C4 and C1-INH testing.
Advertisement
A new case report from Wang and Shen describes the uncommon coexistence of hereditary angioedema (HAE) and chronic spontaneous urticaria (CSU) in a 26-year-old woman, challenging the long-held clinical assumption that these 2 conditions are mutually exclusive.1
HAE is a rare, autosomal dominant disorder characterized by recurrent, nonpruritic angioedema mediated by bradykinin, most commonly due to C1 esterase inhibitor (C1-INH) deficiency caused by mutations in the SERPING1 gene.2 In contrast, CSU is defined by recurrent wheals and pruritus lasting more than 6 weeks without an identifiable trigger and is typically driven by mast cell activation through autoimmune or IgE-mediated mechanisms.
Initial Presentation and Treatment Response
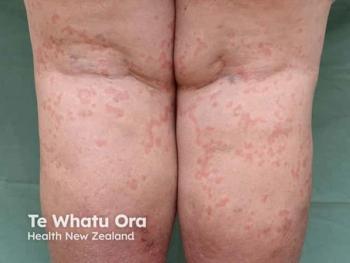
The patient initially presented with a 1-year history of recurrent erythema, wheals, and pruritus affecting the trunk and extremities. These symptoms were consistent with CSU and were refractory to first-line antihistamine therapy. Initiation of omalizumab (300 mg monthly) resulted in significant clinical improvement, including resolution of pruritus and wheals, an increase in the Urticaria Control Test (UCT) score from 7 to 16, and improvement in Dermatology Life Quality Index (DLQI) scores to 5.
Despite adequate control of urticarial symptoms, the patient continued to experience recurrent episodes of angioedema involving the lips and face. These episodes were notable for their lack of associated pruritus or wheals, short duration (30 to 40 minutes), and spontaneous resolution. Importantly, the angioedema episodes occurred independently of CSU disease activity and were not influenced by omalizumab therapy. This atypical presentation prompted further diagnostic evaluation from the clinicians.
Laboratory Findings and Genetic Confirmation
Laboratory investigations revealed significantly reduced complement C4 levels (25 mg/L; reference range 100–400 mg/L) and low C1-INH levels (0.08 g/L; reference range 0.21–0.39 g/L). Subsequent whole exome sequencing identified a heterozygous mutation in the SERPING1 gene (c.991C>A, p.P331T), confirming a diagnosis of type 1 HAE. The patient had no known family history, consistent with the possibility of a sporadic mutation. Given the mild and self-limited nature of her angioedema episodes and minimal impact on quality of life, HAE-specific therapy was not initiated.
Implications for Clinical Practice
This case highlights several important diagnostic and clinical considerations. Historically, the presence of urticaria has been used to exclude HAE, as classic HAE is not associated with wheals. However, this paradigm may lead to missed or delayed diagnoses, particularly when both conditions coexist. Additionally, prodromal manifestations of HAE, such as erythema marginatum, may mimic urticaria and further complicate clinical assessment. In this case, the clear therapeutic response of urticaria to omalizumab, contrasted with persistent angioedema, provided a critical clue prompting further evaluation.
From a pathophysiological standpoint, the coexistence of HAE and CSU raises intriguing questions, as the investigators noted. HAE is driven by dysregulation of the kallikrein–kinin system, resulting in excessive bradykinin production and increased vascular permeability. In contrast, CSU involves mast cell degranulation and histamine release, often associated with autoimmune processes. Emerging evidence suggests potential overlap between these pathways, including complement activation and interactions with the coagulation cascade. Elevated complement components such as C3a and C5a in CSU may promote mast cell activation, while complement dysregulation in HAE may amplify inflammatory responses. These shared mechanisms may partially explain the rare coexistence of both conditions.
Clinically, this report underscores the importance of maintaining a high index of suspicion for HAE in patients with CSU who exhibit atypical angioedema—particularly when episodes are nonpruritic, lack wheals, and do not respond to standard therapies such as antihistamines or omalizumab. Measurement of complement levels, including C4 and C1-INH, should be considered in such cases to avoid diagnostic delay. Overall, greater awareness of this potential overlap may improve diagnostic accuracy, guide targeted therapy, and ultimately enhance patient outcomes.
References
1. Wang H, Shen Y. Case Report: Hereditary Angioedema Accompanied by Chronic Spontaneous Urticaria. Clin Cosmet Investig Dermatol. 2026;19:581977. Published 2026 Mar 23. doi:10.2147/CCID.S581977
2. Reshef A, Kidon M, Leibovich I. The Story of Angioedema: from Quincke to Bradykinin. Clin Rev Allergy Immunol. 2016;51(2):121-139. doi:10.1007/s12016-016-8553-8
Advertisement
Related Content
Advertisement



Latest CME









Advertisement
Advertisement
Trending on Dermatology Times
1
Once-Daily Zasocitinib Rivals Injectable Biologics for Skin Clearance, Phase 3 Data Show
2
FDA Accepts Addition of Bemotrizinol as First New Sunscreen Ingredient in 20 Years
3
Filling the HS Treatment Gap: Ruxolitinib Targets Early-Stage Disease
4
Oral STAT6 Degrader KT-621 Shows Biologic-Comparable Activity in Atopic Dermatitis
5