

|Articles|October 7, 2021
Vitiligo Mechanisms of Melanocyte Death
Author(s)Katie Hobbins
In a recent review, authors examine the different mechanisms of melanocyte death in vitiligo, including genetic predisposition, oxidative stress, and innate vs adaptive immune activation.
Advertisement
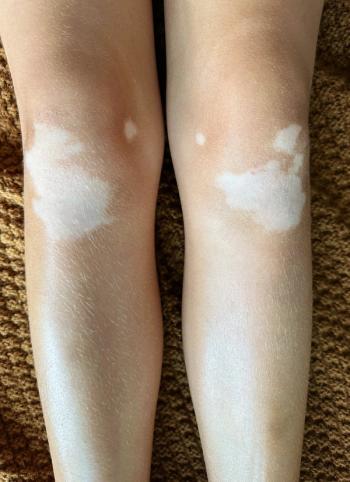
Vitiligo, an autoimmune depigment disease, results from extensive melanocytes destruction through multifactorial causation. In a review on
“Although both segmental vitiligo (SV) and nonsegmental vitiligo (NSV) exhibit melanocyte destruction, whether the destruction of melanocytes in SV is attributed to the autoimmune response or inherent cellular abnormalities remains obscure,” wrote the authors. “Considering that NSV is more prevalent with its pathogenesis more thoroughly studied, we discuss mechanisms of melanocyte death within the scope of NSV… and do not distinguish between ‘NSV’ and ‘vitiligo’.”
The self-responsive immune function, according to the review, directly contributes to the bulk of melanocyte deaths in vitiligo. “The aberrantly heightened innate immunity, type-1-skewed T helper, and incompetent regulatory T cells tip the balance toward autoreaction and CD8+ cytotoxic T lymphocytes finally execute the killing of melanocytes, possibly alarmed by resident memory T cells,” they wrote.
Genetic Predisposition
The concordance of generalized vitiligo in monozygotic twins is approximately 23%, and relatives of patients with vitiligo also hold a 6- to 8-fold risk for the disease compared to the general masses.
An epidemiological study showed that single-nucleotide polymorphisms (SNPs) in multiple risk genes are related to the height of disease. Extensive genome-wide associated studies, according to the review, have uncovered more than 40 susceptibility loci, including MC1R, TYR, IFIH1, CD44, CD80, GZMB, HLA-A, XBP1, CAT, and MTHFR. More than half of the candidate loci are involved in “immunoregulation, T-cell receptor repertoire, and immune cell-associated apoptosis, indicating the critical effect of aberrant immune function in vitiligo pathogenesis,” wrote the authors.
With this knowledge, the list of susceptibility loci could form an important risk evaluation scale to compare against patient gene sequencing and evaluate a parents’ “vitiligo risk score”.
Additionally, a scale of this kind may help optimize diagnosis, prognosis, and more personalized treatment plans.
Oxidative Stress
One of the most crucial initiators in vitiligo is oxidative stress; however, an exact consensus on the etiology remains elusive. Additionally, despite sparse evidence, factors like metabolism could possibly engender melanocyte deaths, according to the review.
Oxidative stress is characterized by “the imbalance of prooxidants and antioxidants,” according to the review. “Oxidative stress in tissue and cell always results from excessive reactive oxygen species (ROS). ROS embody H2O2, hydroxyl radical, hypochlorous acid, and hydroperoxyl radical.”
ROS has proved to be an important second messenger molecule, and in high concentration is implicated in melanocyte death in all aspects, including DNA, lipid, protein, and their metabolites structurally and functionally.
A plausible connection between oxidative stress and deficiency of keratinocyte, melanocyte stem cell, and extracellular microenvironment—based on a growing body of evidence—may help greater inform physician understanding of melanocyte destruction in vitiligo.
Immunological Onslaught
Macrophages, nature killer (NK) cells, innate lymphoid cells (ILC), CTL, regulatory T cells (Treg) and resident memory T (TRM) cells make up nearly every division of the immune system. These cell, and associated cytokines/chemokines, work together to form an immunological barrier and scavenge intrusive pathogens. However, in pathological conditions this barrier recognizes self-antigens and destruct target cells, leading to autoimmune disease.
Considering that the immune system activates in a precise spatio-temporal order, “aberrantly heightened innate immune might play the role of initiator in the derailed immune function,” the authors wrote. “Under stress, [damage-associated molecular patterns molecules]
embodying melanocyte-specific neoantigens like inducible heat shock protein 70 (HSP70i), HMGB1, S100B, and mitochondria DNA are released into the extracellular microenvironment in the form of cell debris and exosome. These DAMPs carrying danger signals further provoke innate immune activation via pattern recognition receptors (PRR) embracing TLRs, RIG-I-like receptors and NLRs.”
As a result, this process could adversely kill melanocytes and ignite adaptive immune through the generation of cytokines/chemokines and antigen-presenting.
Unlike innate immune activation, adaptive immunity is characterized by antigen-specific responses and memorizing antigens. For vitiligo, adaptive immune activation is responsible for specifically killing melanocytes while leaving other cells in the vicinity alone.
“From environmental stimulus to ROS-induced damage and autoimmunity, the program of killing melanocytes is activated in a roughly hierarchical fashion in vitiligo pathogenesis,” the authors concluded.
Reference:
Chen J, Li S, Li C. Mechanisms of melanocyte death in vitiligo. Med Res Rev. 2021;41(2):1138-1166. doi: 10.1002/med.21754
Advertisement
Related Content
Advertisement



Latest CME









Advertisement
Advertisement
Trending on Dermatology Times
1
AbbVie to Acquire Apogee Therapeutics, Expanding AD Pipeline With Zumilokibart
2
Ycanth Phase 3 Trial Dosing Begins for Common Warts in US, Japan
3
Dermatologists and Oncologists Discuss Referral Gaps in Advanced CSCC Care
4
David Rosmarin, MD, on CHE Heterogeneity and Unmet Needs in AD and Vitiligo
5